Aptean Insights
Whether you’re looking for industry insights, product details, upcoming events or our latest news it’s all right here. Browse the categories below or get in touch with one of our experts if you have a question.
Results
Showing1-15of1979
 Press Release Apr 20, 2026Aptean introduces AI platform and AI agents for Business Central On-Premises CustomersAptean unveils AppCentral, an AI platform with 10 AI agents for Business Central on-premises customers—enabling partners to deliver AI without cloud migration and unlock new revenue opportunities.
Press Release Apr 20, 2026Aptean introduces AI platform and AI agents for Business Central On-Premises CustomersAptean unveils AppCentral, an AI platform with 10 AI agents for Business Central on-premises customers—enabling partners to deliver AI without cloud migration and unlock new revenue opportunities.Learn More
 Press Release Apr 7, 2026Aptean Brings Agentic AI to Supply Chain Planning with Logility DemandAI+ on Aptean AppCentralAptean launches Logility DemandAI+ on AppCentral, bringing agentic AI to demand planning for faster decisions, improved forecast accuracy, and smarter supply chains.
Press Release Apr 7, 2026Aptean Brings Agentic AI to Supply Chain Planning with Logility DemandAI+ on Aptean AppCentralAptean launches Logility DemandAI+ on AppCentral, bringing agentic AI to demand planning for faster decisions, improved forecast accuracy, and smarter supply chains.Read More
 Blog Post Mar 25, 2026ERP Migration Mistakes: 5 Critical Areas to AvoidDiscover 5 migration-specific mistakes that derail ERP projects. Learn how legacy system transitions differ from new implementations and avoid costly post-go-live failures.
Blog Post Mar 25, 2026ERP Migration Mistakes: 5 Critical Areas to AvoidDiscover 5 migration-specific mistakes that derail ERP projects. Learn how legacy system transitions differ from new implementations and avoid costly post-go-live failures.Learn More
 Press Release Mar 23, 2026Aptean (Logility) Named a Leader in the 2026 Gartner® Magic Quadrant™ Reports for Supply Chain Planning Solutions: Process industries and Supply Chain Planning Solutions: Discrete IndustriesAptean (Logility) recognized as a Leader in the 2026 Gartner Magic Quadrant for Supply Chain Planning, supporting process and discrete industries.
Press Release Mar 23, 2026Aptean (Logility) Named a Leader in the 2026 Gartner® Magic Quadrant™ Reports for Supply Chain Planning Solutions: Process industries and Supply Chain Planning Solutions: Discrete IndustriesAptean (Logility) recognized as a Leader in the 2026 Gartner Magic Quadrant for Supply Chain Planning, supporting process and discrete industries.Read More
 Blog Post Mar 18, 2026Before and After: How AI in Transportation and Logistics Enables Smarter and Smoother OperationsSee how AI in transportation and logistics transforms daily operations—from maintenance to routing—with real before-and-after scenarios from Jim Endres.
Blog Post Mar 18, 2026Before and After: How AI in Transportation and Logistics Enables Smarter and Smoother OperationsSee how AI in transportation and logistics transforms daily operations—from maintenance to routing—with real before-and-after scenarios from Jim Endres.Learn More
 Success Story Mar 12, 2026Heartland Produce Elevates Warehouse Efficiency With Produce Pro Software from ApteanHeartland Produce improves warehouse efficiency using Produce Pro Software from Aptean, enhancing operations, visibility, and productivity.
Success Story Mar 12, 2026Heartland Produce Elevates Warehouse Efficiency With Produce Pro Software from ApteanHeartland Produce improves warehouse efficiency using Produce Pro Software from Aptean, enhancing operations, visibility, and productivity.Read Story
 Blog Post Mar 12, 2026Comparing Food and Beverage ERP Solutions and Preparing Your Business for ImplementationWith many options to consider—from behemoth generic ERPs to niche sub-industry solutions—how do you identify the right system for your business? Find out how, now.
Blog Post Mar 12, 2026Comparing Food and Beverage ERP Solutions and Preparing Your Business for ImplementationWith many options to consider—from behemoth generic ERPs to niche sub-industry solutions—how do you identify the right system for your business? Find out how, now.Learn More
 Press Release Mar 10, 2026Aptean Launches Its Most Advanced Cloud-Native, AI-Powered Equipment Dealer Management SystemAptean launches its most advanced cloud-native, AI-powered equipment dealer management system, designed to improve efficiency, insights, and dealer operations.
Press Release Mar 10, 2026Aptean Launches Its Most Advanced Cloud-Native, AI-Powered Equipment Dealer Management SystemAptean launches its most advanced cloud-native, AI-powered equipment dealer management system, designed to improve efficiency, insights, and dealer operations.Read More
 Blog Post Feb 25, 2026Logistics Trends To Act On in 2026: What’s Shaping the Industry Right Now2026 won’t be a quiet year for logistics. From AI-driven planning to sustainable transportation and workforce change, see what you should have your eye on.
Blog Post Feb 25, 2026Logistics Trends To Act On in 2026: What’s Shaping the Industry Right Now2026 won’t be a quiet year for logistics. From AI-driven planning to sustainable transportation and workforce change, see what you should have your eye on.Learn More
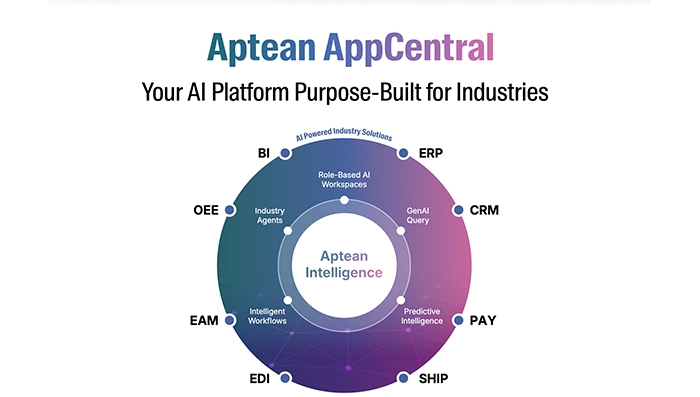 Infographic Feb 19, 2026Aptean AppCentral Your AI Platform Purpose-Built for IndustriesAptean AppCentral is an industry-focused AI platform designed to unify systems, streamline operations, and deliver intelligent insights. Empower your business with scalable, purpose-built AI solutions.
Infographic Feb 19, 2026Aptean AppCentral Your AI Platform Purpose-Built for IndustriesAptean AppCentral is an industry-focused AI platform designed to unify systems, streamline operations, and deliver intelligent insights. Empower your business with scalable, purpose-built AI solutions.Download
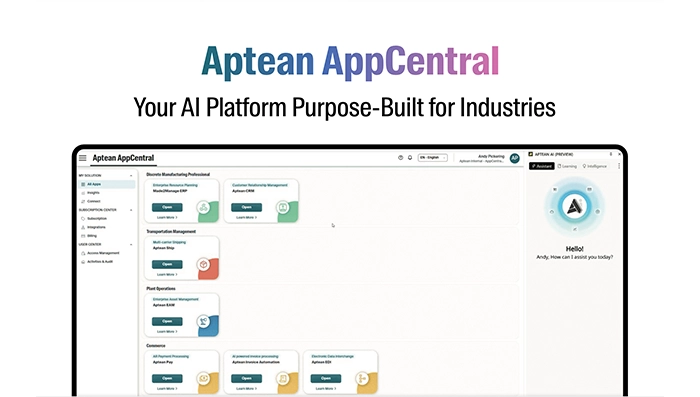 Infographic Feb 18, 2026Aptean AppCentral Your AI Platform Purpose-Built for IndustriesAptean AppCentral is an AI platform purpose-built for industry-specific needs. Connect applications, automate workflows, and drive smarter decisions with scalable, secure AI designed for your business.
Infographic Feb 18, 2026Aptean AppCentral Your AI Platform Purpose-Built for IndustriesAptean AppCentral is an AI platform purpose-built for industry-specific needs. Connect applications, automate workflows, and drive smarter decisions with scalable, secure AI designed for your business.Download
 Blog Post Feb 17, 2026Do You Need Business Route Planning Software? Here's How You Can TellWhether you’re using spreadsheets or basic routing tools, here are the telltale signs that your business needs to switch to business route planning software.
Blog Post Feb 17, 2026Do You Need Business Route Planning Software? Here's How You Can TellWhether you’re using spreadsheets or basic routing tools, here are the telltale signs that your business needs to switch to business route planning software.Learn More
 Blog Post Feb 17, 2026Insights From Microsoft for Building High-Value, Scalable AI ProjectsEduardo Kassner shares Microsoft’s proven framework for choosing, testing and scaling AI projects that deliver real business value.
Blog Post Feb 17, 2026Insights From Microsoft for Building High-Value, Scalable AI ProjectsEduardo Kassner shares Microsoft’s proven framework for choosing, testing and scaling AI projects that deliver real business value.Learn More
 Brochure Feb 17, 2026AppCentral Fashion & Apparel Industry Hub BrochureExplore the AppCentral Fashion & Apparel Industry Hub brochure to learn how our connected ERP platform supports apparel brands with greater visibility, efficiency, and growth.
Brochure Feb 17, 2026AppCentral Fashion & Apparel Industry Hub BrochureExplore the AppCentral Fashion & Apparel Industry Hub brochure to learn how our connected ERP platform supports apparel brands with greater visibility, efficiency, and growth.Download
 Infographic Feb 17, 2026Aptean Intelligence Industry AI That Understands Your BusinessAptean Intelligence delivers industry-specific AI built to understand your business. Unlock smarter insights, streamline operations, and drive better decisions with AI designed for your unique workflows.
Infographic Feb 17, 2026Aptean Intelligence Industry AI That Understands Your BusinessAptean Intelligence delivers industry-specific AI built to understand your business. Unlock smarter insights, streamline operations, and drive better decisions with AI designed for your unique workflows.Download
Ready To Start Transforming Your Business?
We’ve got the specialized industry software to help your organization thrive.
